加拿大医疗器械注册
Medical Device Establishment License MDEL 医疗器械生产许可证
Canadian Medical Device Conformity Assessment System(CMDCAS)加拿大医疗器械合格评定体系
加拿大医疗器械主管部门在实施医疗器械产品注册时,是结合第三方质量管理体系审查,即Ⅱ,Ⅲ和Ⅳ类器械生产企业在申请注册时,需要提交加拿大医疗器械认证认可机构(Canadian Medical Devices Conformity,CMDCAS)认可的第三方机构出具的医疗器械质量管理体系认证证书,即CAN/CSA-ISO 13485体系证书。
加拿大对医疗器械实行上市后监测体系,包括:
1.应用CMDCAS政策,对加拿大市场的医疗器械进行监测;
2.Ⅱ类、Ⅲ类、Ⅳ类医疗器械生产商自始至终实施质量体系;
3. 建立不良事件监测管理体系。
加拿大对医疗器械的分类与欧盟MDD指令中的分类类似,也采用基于风险和规则的分类。在《医疗器械法规》中将医疗器械按风险由低到高分为Ⅰ、Ⅱ、Ⅲ及Ⅳ共四类,具体分类方法依据该法规中的Schedule 1分类规则。如果医疗器械可以被分入多于一个类别,则其分类归类到高的那个类别。Ⅰ类代表最低风险,Ⅳ类代表最高风险。
Ⅰ类:最低风险器械,例如:伤口护理和非外科侵入器械如物理屏障;
Ⅱ类:低风险器械包括隐形眼镜和大多数外科侵入器械;
Ⅲ类:中等风险器械,例如髋关节植入物,葡萄糖监测器和预期可被身体吸收或预期在身体中停留至少30个连续天的外科侵入器械。
Ⅳ类:高风险器械,例如心脏起搏器和用于诊断、控制或纠正一个心血管中枢系统的缺陷的外科侵入器械。
临床要求
加拿大卫生部对医疗器械临床研究的要求请参考GHTF对医疗器械临床研究的要求。加拿大一般认可FDA临床数据。
市场准入
Health Canada要求医疗器械生产企业满足质量体系要求,并且是由Canadian Medical Device Conformity Assessment System(CMDCAS)认可的公告机构的质量体系认证。Ⅱ,Ⅲ和Ⅳ类医疗器械的生产商的质量管理体系必由Health Canada 认可的公告机构通过CAN/CSA ISO 13485质量体系审核。通过质量体系认证之后,向Health Canada申请产品注册Medical Device License,如果持证18个月后仍没有获得加拿大产品注册证,则质量体系证书将被撤销。
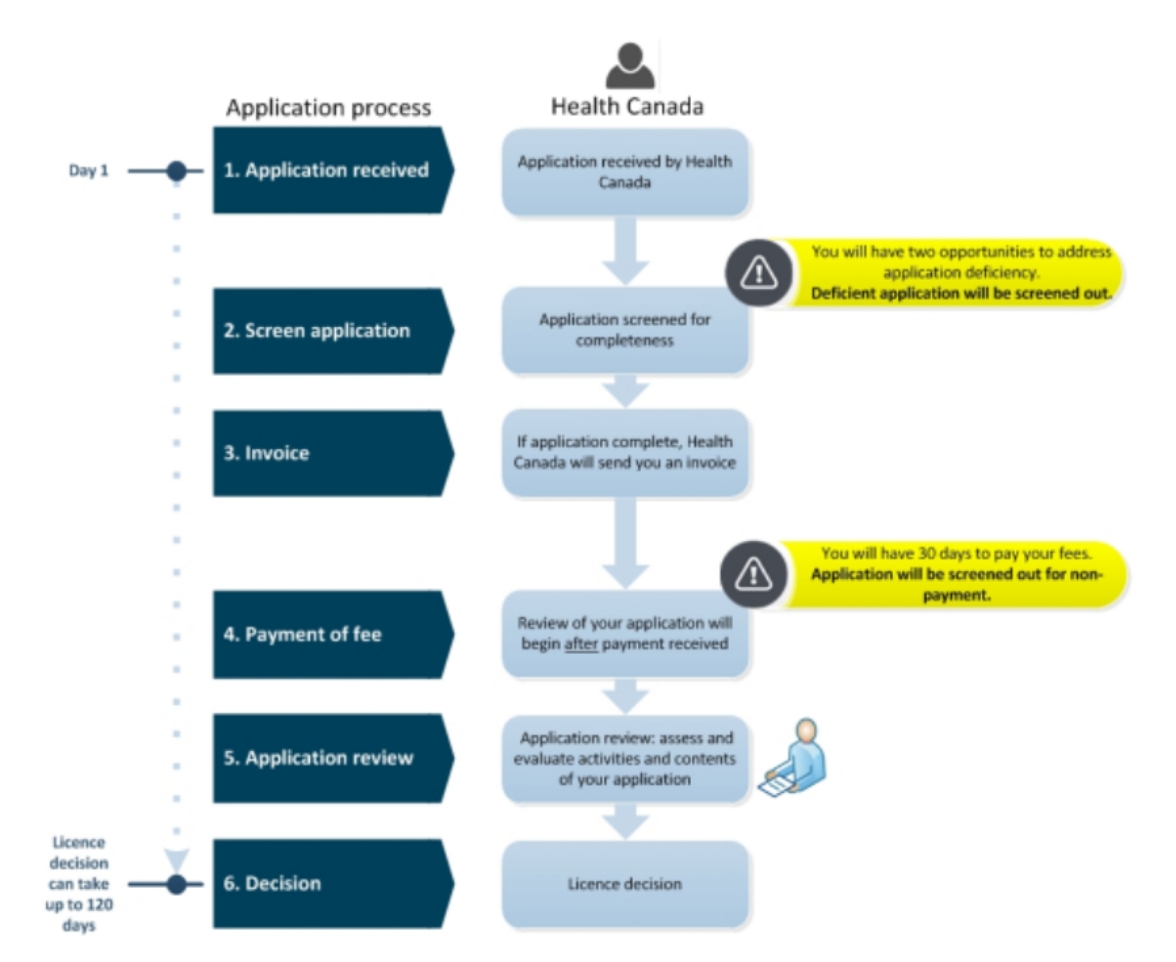
根据产品的风险分类和特性,按照Ⅱ,Ⅲ和Ⅳ类的提交文件清单进行准备,拟制相应的申请文件提交给加拿大健康部,一般会在1~2个月提出审核意见,在3-6个月内通过许可。加拿大的审核基本分为4个步骤,每个步骤都可能提出问题,并告知回复问题的期限,一般为10天到30天之间。每个环节只有2次回复的机会,否则将会拒绝该注册。通过认证后,加拿大健康部会电邮一份电子版的证书和一份纸质版的证书,每年需要提交证书继续保留或取消的申请,并支付继续保留的年费。如果在加拿大的年销售额不超过2万加币,可以申请费用优惠。按照证书的数目来支付年费。
Health Canada加拿大卫生部颁发证书,并在数据库上更新器械的证书号。加拿大证书无有效期,每一年需要但是如果涉及到产品变更的话,需要提交变更申请,证书号只会更新发证日期,证书号不改变。
质量监管
加拿大健康卫生部是加拿大医疗器械主管部门,其质量体系要求基于《加拿大医疗器械法》和ISO13485:2003。质量体系证书(CAN/CSA-ISO13485:2003)颁发由加拿大认证认可机构(CMDCAS,CanadianMedical Devices Conformity)认可的第三方机构出具,证书有效期3年,收取费用同欧盟一样随认证机构、制造商的产品风险级别的高低、技术文件、生产工艺的复杂程度、企业的人员数和规模等不同而不同。需要注意的是,当制造商的质量体系证书更新时,应在30天内通知加拿大健康卫生部。
加拿大医疗器械产品的上市后管理包括:应用CMDCAS政策进行检测;Ⅱ类, Ⅲ类, Ⅳ类产品始终实行质量体系(QS);建立不良事件监督管理体系。
Ⅱ,Ⅲ和Ⅳ类医疗器械的生产商的质量管理体系必由Health Canada(加拿大卫生部) 认可的公告机构通过CAN/CSA ISO 13485质量体系审核。通过质量体系认证之后,向Health Canada申请产品注册MDL,如果持证18个月后仍没有获得加拿大产品注册证,则质量体系证书将被撤销。
根据产品的风险分类和特性,按照Ⅱ,Ⅲ和Ⅳ类的提交文件清单进行准备,拟制相应的申请文件提交给加拿大健康部,一般会在1~2个月提出审核意见,在3-6个月内通过许可。加拿大的审核基本分为4个步骤,每个步骤都可能提出问题,并告知回复问题的期限,一般为10天到30天之间。每个环节只有2次回复的机会,否则将会拒绝该注册。通过认证后,加拿大健康部会电邮一份电子版的证书和一份纸质版的证书,每年需要提交证书继续保留或取消的申请,并支付继续保留的年费。如果在加拿大的年销售额不超过2万加币,可以申请费用优惠。按照证书的数目来支付年费。
Health Canada加拿大卫生部颁发证书,并在数据库上更新器械的证书号。加拿大证书无有效期,每一年需要但是如果涉及到产品变更的话,需要提交变更申请,证书号只会更新发证日期,证书号不改变。
HC流程说明流程说明
备注
1、根据加拿大法规定义,判定该设备是否为医疗器械 参考加拿大法规SOR/98-282
2、完成加拿大ISO13485的质量体系认证,才能开始做产品注册。 参考加拿大法规SOR/98-282
3、对产品进行分类,分为Ⅰ,Ⅱ,Ⅲ和Ⅳ类
一般只有Ⅱ,Ⅲ和Ⅳ类需要注册。
注册费用参考指南:Fees for the Review of Medical Device License Applications
4、对产品的风险分类后,根据类别提交文件。根据类别,指南对要求对应的文件提交清单。
5.通过认证后,加拿大健康部会电邮一份电子版的证书和一份纸质版的证书。
证书永久生效,只针对当时认证的产品。
6.结束后,如果产品涉及到变更等,需要提交变更申请。流程重新开始。

所有在加拿大境内进口和销售医疗器械的法人和自然人以及I类医疗器械的制造商都需要获得机构许可(Medical Device establishment Licence, MDEL)。II、III和IV类医疗器械制造商、医疗机构和零售上商不需申请获得MDEL。
I 类医疗器械的制造商无需产品注册,但II、III和IV类医疗器械的制造商须在申请MDEL之前先获得产品注册许可证(Medical Device License, MDL).
(1)营业许可证的申请应提交以下材料:
a) 营业单位的名称/地址;
b) 作为情况联系人的营业单位代表的名称、职位及电话号码;
c) 介绍营业单位为进口商或销售商或二者兼有;
d) 进口或销售的医疗器械制造者的名称地址;
e) 针对每一制造者,列在卫生部所指定的专家库中与进口或销售医疗专家;
f) 针对每一制造者,进口或销售的器械的分类;
g) 营业单位的高级官员所作的声明,声明营业单位建立并保持与分销记录、投诉处理和召回相关的形成文件的程序;
h) 若营业单位进口医疗器械,见其高级官员声明遵守强制采取报告制度;
I) 当营业单位进口或销售II,III,IV类医疗器械,高层官员声明建立文件化程序以控制这些器械处理、贮存、交付、安装、纠正措施和服务;
j) 在加拿大境内实施(g)至(I)款的各个地址。
所有已获得MDEL的机构都需在每年4月1日之前提交年度审评申请,以延续有效MDEL.对于已获得MDEL但生产活动还未满一年的制造商而言,MDEL审评费用可延期至第一个日历年结束时缴纳。
五、CMDCAS审核点
对I类医疗器械(包括IVD)没有质量体系要求,并豁免产品注册许可。
加拿大医疗器械法规(Canadian Medical Device Regulations, CMDR)不要求进口商或分销商进行质量体系注册。
加拿大的现行质量体系标准为CAN/CSA-ISO 13485-03(R2013)。
II类医疗器械制造商应符合中除设计以外的要求;III和IV类器械应符合包括设计在内的所有CAN/CSA-ISO 13485-03(R2013)要求。
需要提交加拿大医疗器械认证认可机构(Canadian Medical Devices Conformity,CMDCAS)认可的第三方机构出具的医疗器械质量管理体系认证证书,即CAN/CSA-ISO 13485体系证书。
做这个体系认证的机构是哪些? TUV(四家), PS, BSI, BV, ITS, NSAI ,SGS, SAI, UL LLC ,DQS, G-MED 全球一共十五家